
Markers for Lr37-Yr17-Sr38
A long chromosomal fragment (25-38 cM) containing three rust resistance genes was translocated between the short arms ofTriticum ventricosum 2NS and the bread wheat chromosome 2AS (1). This segment includes three disease resistance genes: Lr37, Yr17 and Sr38 conferring resistance to leaf rust (Puccinia triticina Eriks), stripe rust (Puccinia striiformis West. f. sp. tritici) and stem rust (Puccinia graminis Pers. f.sp. tritici Eriks. & E. Henn.) respectively.
The 2NS fragment was first introgressed into wheat cultivar VPM1 (2) and later it was transferred to other commercial cultivars like Madsen and Thatcher (3,4,5,6). Recently, Helguera et al. found thatLr37 was not functional in Anza-Lr37, suggesting the presence of a supressor factor (7).
Rust races virulent to Lr37 and Yr17 resistance genes have been already identified (5). However, these genes still provide resistance to a wide range of races and can be used in combination with other rust resistance genes.
Methods
Helguera et al. (7) developed a TaqMan®-based PCR marker for this fragment, but there are also some other conventional PCR markers available. The 2NS chromosome segment does not recombine with the bread wheat chromosomes, and therefore the three resistance genes are transferred together and are completely linked to markers developed within the 2NS segment. However, occasional recombination events cannot be ruled out and the final products should be tested with the pathogen to confirm the introgression of resistance genes. Until now, we have not observed a single recombination event between the markers and the resistance genes.
TaqMan® assay for Lr37 (7):
A PCR assay was developed using TaqMan® technology as a high-throughput alternative for selection of the 2NS/2AS translocation in large segregating populations in breeding programs that have access to real time PCR equipment. This technology uses real time fluorogenic detection of PCR products and therefore does not require gels to visualize the PCR products, facilitating automation of the MAS process.
Reactions were performed using a system composed by a pair of primers (VENTRIUP - LN2) with the probe W2NS-55T. The probe included a single point mutation at bp 17 that differentiated the 2NS allele from the 2A, 2B and 2D alleles. The probe was labeled with the fluorescent reporter dye 6-carboxy-fluorescein (FAM) at the 5' end and the quencher 6-carboxy-N,N,N',N'-tetramethyl-rhodamine (TAMRA) at the 3' end. When the target sequence is being amplified, the 5' nuclease activity of the Taq DNA polymerase cleaves the fluorogenic probe included into the PCR reaction. The physical separation of the fluorescent reporter dye from the quencher causes an increase in its fluorescence intensity that is detected in "real time" by the quantitative PCR equipment.
Primers:
VENTRIUP 5'- AGGGGCTACTGACCAAGGCT -3'
LN2 5'- TGCAGCTACAGCAGTATGTACACAAAA -3'
Probe:
W2NS-55T 5'- FAM-TGTTTGGTTCCTATCTCCTTCCTGGTCCTG-TAMRA -3'
Final concentrations of the products used in the PCR reaction:
- primers: 0.4 µM each
- probe: 0.08 µM
- 1X TaqMan® Universal PCR Mastermix (Applied Biosystems) :
- 10 mM Tris-HCl (pH 8.3)
- 50 mM KCl
- 5 mM MgCl2
- 2.5 mM deoxynucleotide triphosphates
- 0.625 U AmpliTaq Gold DNA polymerase per reaction
- 0.25 U AmpErase UNG per reaction
- DNA input: 200 ng/reaction
Final volume: 25 µL
PCR conditions
The samples were placed in 96 well plates and amplified in an ABI Prism 7700 Sequence Detection System (Applied Biosystems).
- 2 min at 50°C
- 10 min at 95°C
- Amplification step (40 cycles)
- 95°C 15 sec
- 60°C 60 sec
Expected products:
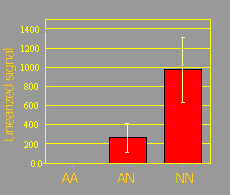
The figure below shows the amplification signal obtained from 24 individuals with known AA, AN, and NN genotypes using TaqMan® system. The analysis of variance for the amplification signal showed significant differences among genotypes (P= 0.002). The response was linear as evidenced by a significant linear response (P=0.006) and a non-significant quadratic response (P=0.48).
Conventional PCR markers
There are two markers available for detecting the presence of the fragment containing the genes Lr37-Yr17-Sr38. The first is a PCR marker, dominant for the 2NS segment and that can be converted into a codominant marker. The second marker is a CAPS (cleavage amplified polymorphic sequence) marker. This CAPS marker requires an extra step of digestion with a restriction enzyme after the PCR amplification, but has two advantages, it is codominant and can be used with DNA of lower quality than the PCR marker.
PCR marker specific for the N genome:
The pair of primers VENTRIUP-LN2 was designed to amplify preferentially the N-allele of Xcmwg682. The 3' end from the left primer VENTRIUP matches a point mutation at position 46 that differentiates the N-allele from the other three wheat alleles, while the 3' end of the right primer LN2 matches a TT indel at positions 261-262 that is specific for the N and A-alleles.
By adding a third primer (Yr17neg-F), designed to target the A-genome difference of the ventruip gene, an agarose codominant marker was developed at Oregon State University (Adam Heesacker, personal communication).
Primers:
VENTRIUP 5'- AGGGGCTACTGACCAAGGCT -3'
LN2 5'- TGCAGCTACAGCAGTATGTACACAAAA -3'
Yr17neg-F 5'- GATCCATGACGCGCATTTG -3'
Final concentrations of the products used in the PCR reaction:
- 1X Taq buffer (Mg2+ free)
- dNTP: 200 µM each
- MgCl2: 1.5 mM
- primers: 0.2 µM each
- Taq polymerase: 1.0 U
- DNA input: 120 ng/reaction
Final volume: 25 µL
PCR conditions (times for each step might vary according to the Thermocycler model):
- Denaturing step: 94°C, 45 sec
- Amplification step (30 cycles)
- 94°C 45 sec
- 65°C 30 sec
- 72°C 60 sec
- Extension step: 72°C 7 min
Expected products:
The two-primers protocol is expected to amplify a 259-bp fragment in plants carrying one or two doses of the 2NS translocation.
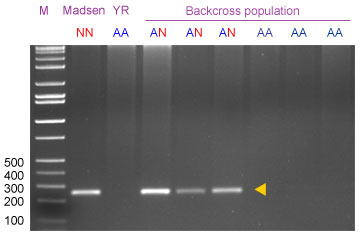
With the three-primers protocol, resolved on an agarose gel, there are two possible products: An upper band (260 bp) corresponding to the original dominant product of VENTRIUP/LN2, and a lower band (163 bp) due to amplification with Yr17neg/LN2 indicating absence of Yr17 .
CAPS marker specific for the A genome:
The 259-bp PCR product from primers VENTRIUP-LN2 is a dominant marker and therefore cannot differentiate heterozygous from homozygous 2NS individuals. A marker to differentiate these two classes is a desirable tool for selection of 2NS homozygous plants in F2 segregating populations or after self-pollination of the heterozygous BC plants from the last cycle of a backcrossing program. One way to differentiate the homozygous 2NS plants from the heterozygous is by the absence of the 2A-allele in the homozygous 2NS plants. A CAPS marker for the 2A-allele was developed to complement the 2N-allele specific primers: primers URIC and LN2 amplify preferentially the N and A-alleles of Xcmwg682. These two alleles can then be differentiated by the presence of a Dpn II restriction site at position 110 that is present in the A and B genomes and is absent in the D and N genomes (see sequences).
Primers:
URIC 5'- GGTCGCCCTGGCTTGCACCT -3'
LN2 5'- TGCAGCTACAGCAGTATGTACACAAAA -3'
Final concentrations of the products used in the PCR reaction:
The reagents and concentrations are the same as for the previous set of primers.
PCR conditions (times for each step might vary according to the Thermocycler model):
- Denaturing step: 94°C, 45 sec
- Amplification step (38 cycles)
- 94°C 45 sec
- 64°C 30 sec
- 72°C 60 min
- Extension step: 72°C 7 min
Digestion with Dpn II:
Following amplification with CAPS primers, 5µL of the PCR reaction were digested with restriction enzyme Dpn II by adding 5 U of enzyme to the PCR product. Samples were separated by electrophoresis in 2% agarose gel and visualized using ethidium bromide and UV light.
Expected products:
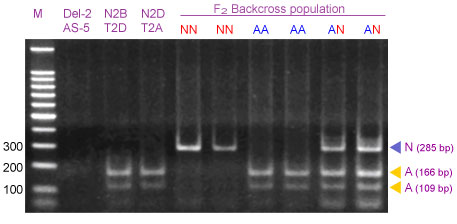
Amplification of wheat genomic DNA from heterozygous 2AS/2NS plants with CAPS primers URIC and LN2 amplify two fragments of 285-bp (N-allele) and 275-bp (A-allele). Although the size difference between these two fragments is small, they can be clearly separated in polyacrylamide gels as well as in 4% Metaphor agarose gels. However, both alleles can be better differentiated after digestion with Dpn II: an undigested 285-bp band corresponds to the N genome PCR product and the 166 and 109-bp fragments corresponded to the digested PCR product of the A genome.
References
1. Cytogenetic studies in wheat XIV. Location of rust resistance genes in VPM1 and their genetic linkage with other disease resistance genes in chromosome 2A. Bariana HS, McIntosh RA. In: Genome, 1993, 36: 476-482. DOI:10.1139/g93-065
2. Obtention des bles tendres resistants au pietin-verse par croisements interspecifiques bles X Aegilops. Maia N. In: C. R. Acad. Agric. Fr., 1967, 53: 149-154.
3. The genetic analysis of two interspecific sources of leaf rust resistance and their effect on the quality of common wheat. Dyck PL, Lukow OM. In: Canadian Journal of Plant Science, 1988, 68: 633-639. DOI:10.4141/cjps88-076
4. McIntosh RA, Wellings CR, Park RF. In: Wheat Rusts, an Atlas of Resistance Genes Melbourne, Australia, CSIRO, 1995.
5. Identification of molecular markers for the detection of the yellow rust resistance gene Yr17 in wheat. Robert O, Abelard C, Dedryver F. In: Molecular Breeding, 1999, 5:167-175. DOI:10.1023/A:1009672021411
6. Resistance gene analogs within an introgressed chromosomal segment derived from Triticum ventricosum that confers resistance to nematode and rust pathogens in wheat. Seah S, Spielmeyer W, Jahier J, Sivasithamparam K, Lagudah ES. In:Molecular and Plant Microbe Interactions, 2000, 13: 334-341. DOI:10.1094/MPMI.2000.13.3.334
7. PCR assays for the Lr37-Yr17-Sr38 cluster of rust resistance genes and their use to develop isogenic hard red spring wheat lines Helguera M, Khan IA, Kolmer J, Lijavetzky D, Zhong-qi L, Dubcovsky J. In: Crop Science, 2003, 43:1839-1847. DOI:10.2135/cropsci2003.1839